Molecular Modeling

Molecular modeling is used within the MIME team as a computational microscope to understand and decipher the interactions between the different constituents of the ExtraCellular Matrix (ECM) at the atomic level. Taking into account the great diversity of actors and the different degrees of complexity of ECM, we use usual approaches of molecular mechanics, molecular dynamics and molecular docking. We also carry out dedicated methodological developments both for the implementation of new calculation techniques and for the analysis of the numerical big data generated. Using molecular modeling, we (i) characterize the structure/activity relationship of ECM degradation products, (ii) study the interactions between matrikines and proteins of the ECM, and (iii) develop methodologies dedicated to the exploration and analysis of biological systems (see below).
Visualization and Extended Reality

The growing evolution of computing systems and HPC towards exascale level has created an important need for specific tools that would assist the scientist in the visualization of increasingly complex and huge molecular data, such as those encountered in the ECM. For this reason, new representation modes must be added to the existing ones, to allow the 3D visualization of all simulated data. In addition, virtual reality also provides innovative solutions to issues related to visualization and complex simulations control. Moreover, combining stereoscopic rendering, very useful in molecular visualization, and haptic modalities, particularly suitable for interacting with molecules, we are developing tools for complete immersive visualization and new modes of representation. These allow the analysis of the cell-matrix interface by adapting to the complex multi-domain macromolecules such as matrix proteins (thrombospondins, elastin, collagens, etc.) or saccharides (glycans and glycosaminoglycans, see picture).
Image Processing
MIME’s image analysis is focused on both qualitative and quantitative exploitation of images from ECM,and is interested in the statistical inference associated with them. This work aim at developing and implementing innovative imaging and image analysis methods to answer questions related to ECM remodeling during aging and aged-related diseases such as metabolic disorders, vascular diseases, and/or tumor progression
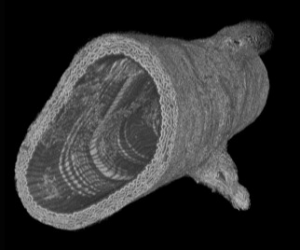 One of our approaches consists in imaging matrix structures at the sub-micrometric scale using synchrotron X-ray micro- tomography (say micro-CT with a synchrotron source). A series of images from murine aortas were obtained on the Diamond Light Source synchrotron (Didcot, UK) in collaboration with Dr. M.J. Sherratt from the University of Manchester. This work has demonstrated the potential of the developed methodology. It was continued with new acquisitions made at the ANATOMIX beamline from SOLEIL synchrotron (Saclay, FR). Image processing and analysis is underway, notably by including artificial intelligence approaches to extract the relevant information from these big data images.
One of our approaches consists in imaging matrix structures at the sub-micrometric scale using synchrotron X-ray micro- tomography (say micro-CT with a synchrotron source). A series of images from murine aortas were obtained on the Diamond Light Source synchrotron (Didcot, UK) in collaboration with Dr. M.J. Sherratt from the University of Manchester. This work has demonstrated the potential of the developed methodology. It was continued with new acquisitions made at the ANATOMIX beamline from SOLEIL synchrotron (Saclay, FR). Image processing and analysis is underway, notably by including artificial intelligence approaches to extract the relevant information from these big data images.
Biospectroscopies and probes
The spectroscopic work carried out in our group aims to characterize the structures and conformational modifications of the ECM molecules. The methods used are electronic and
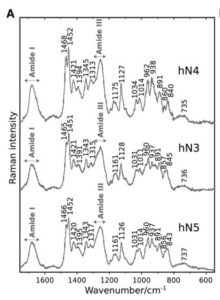
vibrational circular dichroism (ECD, VCD) spectroscopy, infrared absorption (IR) and Raman scattering spectroscopies. The processing and analysis of the recorded spectra provide essential experimental information used to guide modellers when they build molecular models from their theoretical structural calculations.
This approaches have been recently applied to nonapeptides derived from elastin (see Figure ) and published in Biophysical Journal.
Methodological developments
To answer the questions raised by teams 1 and 2, the MIME team develops and proposes methodological and technical solutions to address their concerns. These developments are made at the biology/physics/computer science interface and are, initially, matrix-oriented. The major advances made recently and/or still under developments are the following:
AMIDE (since 2012) Workflow allowing to use the capabilities of HPC (High Performance Computing) computers such as ROMEO (romeo.univ-reims.fr) to perform so-called reverse docking, i.e. allowing a small molecule (for example natural substance) or a small peptide (for example matrikine) to target several tens or even hundreds of proteins in a single pass and bring out the best hits. We are currently adding deep learning methodologies to it and try to increase the performance of the program. We have developed the concept of double docking allowing to ”dock” a database of ligands (several hundreds of thousands) on a database of proteins (up to one hundred). The analysis tools are still under development.
As part of the ANR-Flash COVID, a consortium led by our chemist colleagues from the Institute of Molecular Chemistry from our University used this approach to double screen natural substances on a set of SARS-COV-2 proteins and some of their conformations. Analyzes are in progress.
DARMA (2015 – 2017) Study of the processing of aortic images by recognition of arterial cuts and statistical processing of the information contained. This development is at the origin of the work leading to the following DOREMI project.
DOREMI (since 2018) Based on previous developments, the implementation of a workflow allowing the identification, quantification and prediction of arterial alterations at different scales is developed. This includes the implementation from in vivo protocols, sample preparation methods, acquisitions on synchrotrons (DLS or SOLEIL) up to developments for image processing and interpretation.
DURABIN (since 2016) Development of a mesoscopic demonstrator of the extracellular matrix allowing the simulation of several hundreds of ECM macromolecules, based on the use of physical engines from video games in a virtual reality environment combining a set of programs. All these tasks are integrated into the UnityMol X software environment, supported by the laboratory of Dr. M. Baaden in Paris.

A database of three-dimensional structures of matrix protein fragmentsdefined as primitives for mesoscopic simulation is under development. A PhD’ thesis is currently in progress to integrate elastic peptides and tropoelastin (monomer of elastin) in the database. Virtual reality support to control the environment is carried out and the integration of Deep Learning approaches to the analysis of long-duration simulations is underway.
LIMONADA (since 2017) Creation of a database dedicated to lipids and different types of membranes. LIMONADA is an open database that allows to choose, in a context of modeling and simulations, the nature of the lipids and to set up these simulations. Getting realistic biological membrane composition, the possibility to define the asymmetry of the leaflets and the future integration of physico-chemical and biophysical parameters are the important points of this database. The development of this database will be continued to integrate new lipids from lipidomics studies and to set up membrane systems specific to MEDyC studies. LIMONADA is accessible via limonada.univ-reims.fr and is the result of a collaboration with the teams of Dr. L. Lins from the University of Liège (Belgium) and Prof. C. Sarazin from the University of Amiens.
Umbrella Visualization (since 2016) Structural and dynamic characterization by dedicated visualization of the behavior of glycans and glycosaminoglycans of glycoproteins and proteoglycans and by analyzing the possible consequences of the “shadow” cast by the sugar on the protein surface. The whole is developed in the UnityMol X environment and will possibly be integrated in DURABIN.